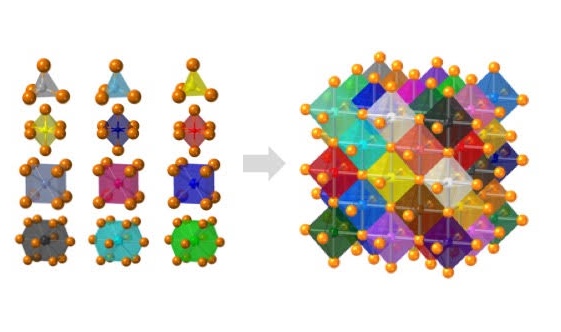
 Crystals with high configurational entropy, engineered through chemical formulation, exhibit unique composition-structure-property combinations that are absent when chemical order prevails. These high-entropy materials follow unexpected crystal chemistry rules and hold promise for new functional properties. IRG2 endeavors to identify and understand these rules through an integrative effort linking theory, synthesis, characterization, and computation. The hypothesis driving this research is that high configurational entropy leads to high solubility for atoms in “misfit” local environments, which produces a spectrum of local energies and disordered geometries that collectively generate new macroscopic responses. The IRG seeks to understand how local structures relate to specific formulations and how short-range disorder couples over longer length scales. With this understanding, we will uncover the predictive rules for high-entropy crystal chemistry.
Crystals with high configurational entropy, engineered through chemical formulation, exhibit unique composition-structure-property combinations that are absent when chemical order prevails. These high-entropy materials follow unexpected crystal chemistry rules and hold promise for new functional properties. IRG2 endeavors to identify and understand these rules through an integrative effort linking theory, synthesis, characterization, and computation. The hypothesis driving this research is that high configurational entropy leads to high solubility for atoms in “misfit” local environments, which produces a spectrum of local energies and disordered geometries that collectively generate new macroscopic responses. The IRG seeks to understand how local structures relate to specific formulations and how short-range disorder couples over longer length scales. With this understanding, we will uncover the predictive rules for high-entropy crystal chemistry.
IRG2 is organized into four interwoven thrusts. Three of these are property-driven and endeavor to understand:
- How electronic and ionic transport can be maximized in high-entropy perovskites and fluorites
- How local distortions can influence global symmetry and polar ordering in high-entropy perovskites with disorder on both cation sublattices
- How electron correlation and magnetism manifest in high-entropy rock salts and pyrochlores.
In each case we explore the limits of misfit cations in a parent high-symmetry structure, where the misfits are chosen so as to influence property evolution. An overarching multiscale theory and modeling effort couples to machine learning to predict structure, defect chemistry, and properties in all materials of interest. At small length scales first-principles calculations model and predict the entire landscape of local distortions in particular formulations and link them to local properties such as ion migration barrier, defect formation energy, band structure, and polarizability. First-principles data feeds phase-field and stochastic models at higher length scales to connect formulation, local structure, electronic structure, and crystal structure to cooperative responses, and microstructures.