
The Center for Nanoscale Science supports collaborative, interdisciplinary research efforts on nanoscale materials. Principal research activities are organized into two interdisciplinary research groups: 2D Polar Metals & Heterostructures and Crystalline Oxides with High Entropy. Center-initiated programs encourage collaborative partnerships with science museums and non-R1 universities as well as engagement in outreach, education, and workforce development initiatives.
2D Polar Metals and Heterostructures
IRG1 2D Polar Metals and Heterostructures pursues the promise of a new materials platform that stabilizes a diverse array of two-dimensional polar metals and enables their integration into ground-breaking optically and electronically active heterostructures.
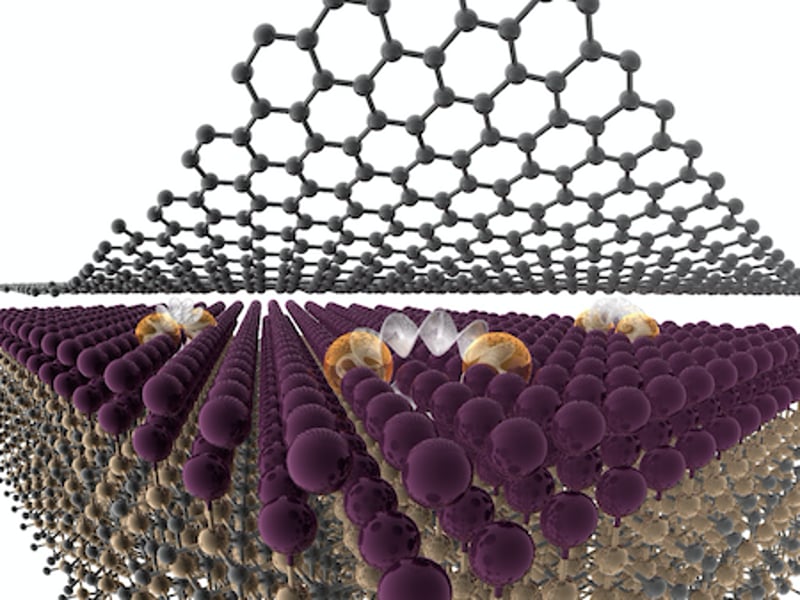
Metals and alloys sit at the heart of materials research, but their susceptibility to surface oxidation has impeded their investigation in atomically thin form or as pristine surfaces exposed to the ambient environment. Thus, metals are generally not considered to be electrostatically gateable, rarely strongly polar, and typically not straightforward constituents of complex quantum hetero-structures due to interfacial reactions. IRG1 surmounts these challenges and opens up new areas of fundamental science and application for metals and their alloys through in-situ encapsulation and heterostructure formation that takes advantage of the protected high-energy interface underneath epitaxial graphene and exploits a self-healing effect that yields air-stable atomically thin crystalline metals that are also polar, with exceptional nonlinear optical response and intriguing potential for impacts in quantum devices and biosensing.
The IRG converges expertise in synthesis, optics and spectroscopy, transport, spintronics, device engineering, biosensing, theory and data-driven computation to exploit the unique opportunities in fundamental science and application afforded by air-stable crystalline 2D metals and alloys. These efforts will be accelerated by predictive computation to guide synthesis and application within the expansive compositional design space that CHet endows, and will open new routes to Quantum Leap, enable new sensing modalities for elucidating the Rules of Life, and provide an intriguing venue to Harness the Data Revolution. The team's efforts are organized around quantum and optical property domains, tied together by a central thrust in synthesis of novel structures, compositions and heterostructures of air-stable polar 2D metals.


Crystalline Oxides with High Entropy
IRG2 explores a new class of crystalline oxides enabled by configurational entropy which offers exciting functional properties and pathways to new basic science.
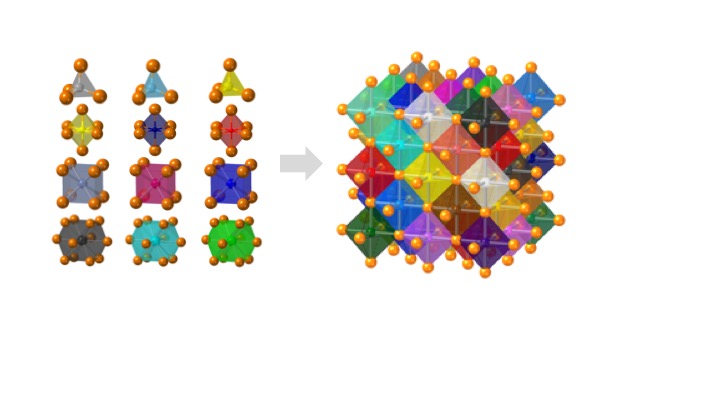
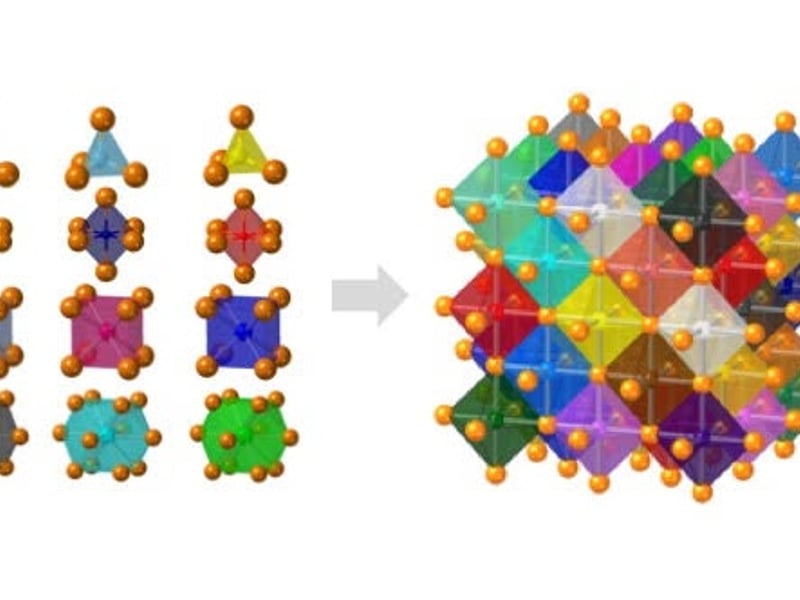
Crystals with high configurational entropy, engineered through chemical formulation, exhibit unique composition-structure-property combinations that are absent when chemical order prevails. These high-entropy materials follow unexpected crystal chemistry rules and hold promise for new functional properties. IRG2 endeavors to identify and understand these rules through an integrative effort linking theory, synthesis, characterization, and computation. The hypothesis driving this research is that high configurational entropy leads to high solubility for atoms in “misfit” local environments, which produces a spectrum of local energies and disordered geometries that collectively generate new macroscopic responses. The IRG seeks to understand how local structures relate to specific formulations and how short-range disorder couples over longer length scales. With this understanding, we will uncover the predictive rules for high-entropy crystal chemistry.
IRG2 is organized into four interwoven thrusts. Three of these are property-driven and endeavor to understand:
How electronic and ionic transport can be maximized in high-entropy perovskites and fluorites
How local distortions can influence global symmetry and polar ordering in high-entropy perovskites with disorder on both cation sublattices
How electron correlation and magnetism manifest in high-entropy rock salts and pyrochlores.
In each case we explore the limits of misfit cations in a parent high-symmetry structure, where the misfits are chosen so as to influence property evolution. An overarching multiscale theory and modeling effort couples to machine learning to predict structure, defect chemistry, and properties in all materials of interest. At small length scales first-principles calculations model and predict the entire landscape of local distortions in particular formulations and link them to local properties such as ion migration barrier, defect formation energy, band structure, and polarizability. First-principles data feeds phase-field and stochastic models at higher length scales to connect formulation, local structure, electronic structure, and crystal structure to cooperative responses, and microstructures.







